

概要
Structure-Based Drug Design
Structure-Based Drug Design (SBDD)は、標的タンパク質や受容体の立体構造情報に基づいた分子設計法です。MOEのSBDDに関連する機能として、表面のもつ特性解析や化合物のドッキングシミュレーション、結合部位での対話的な分子設計、フラグメントを用いた分子設計によって新規リガンド候補を探索・提案します。また、タンパク質-リガンド相互作用解析機能を組み合わせて、妥当な候補構造の絞り込みや重要な相互作用の検出が可能です。
 ドッキングシミュレーション
ドッキングシミュレーション
標的タンパク質の活性部位において、化合物の配置を探索し、化合物の安定な結合配座を予測します。評価関数には、静電、ファンデルワールス、溶媒和の各相互作用エネルギーと、接触表面積に基づいた関数、電子密度マップとの重なりに基づいた関数などから選択できます。タンパク質構造を可動にしたInduced-Fitドッキングにも対応しています。タンパク質の活性部位における化合物の配座の探索のために、以下の手法が搭載されています。
【MOEに搭載されているドッキング手法】
- 標準ドッキング
複合体構造のリガンド周辺やポケット探索で得られたアルファ球を基準に化合物を配置します。
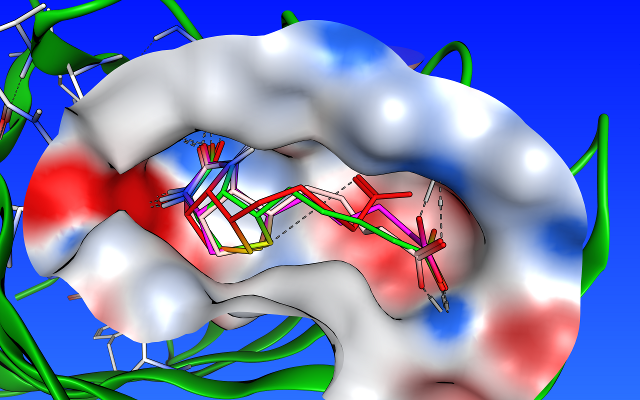
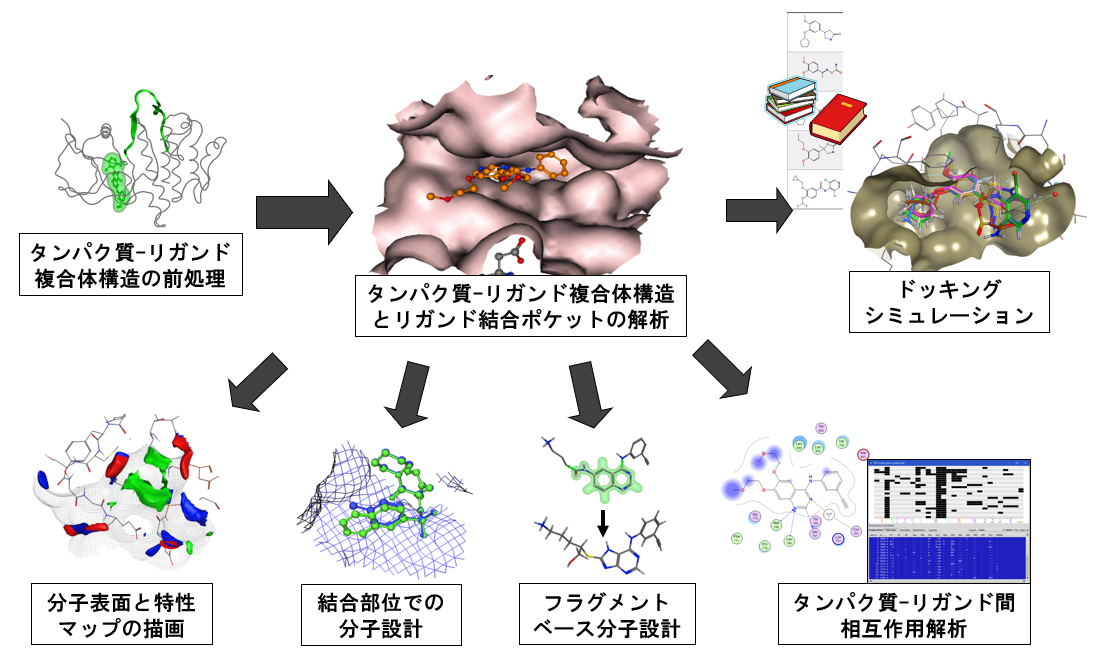
- テンプレートドッキング
標的タンパク質とリガンドの共結晶構造におけるリガンドの部分構造や類似原子を基準として化合物を配置します。
カテコール構造をテンプレートとしたドッキングシミュレーション
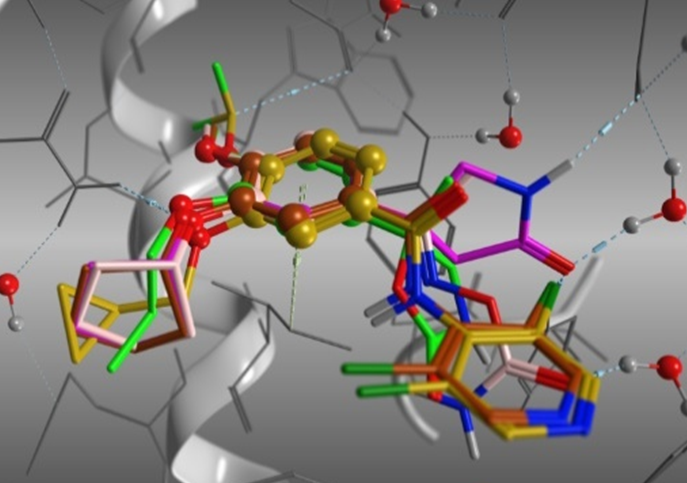
- Covalent Docking
活性部位に化合物を配置し、化学反応に従って標的タンパク質と共有結合させます。化学反応式の編集や追加、標的タンパク質の反応部位の指定ができます。
マイケル付加反応によりリガンドと共有結合的にドッキング
- 電子密度フィッティング
電子密度と重なるように化合物の配置と構造最適化計算を行います。
- タンパク質(核酸)-タンパク質ドッキング
タンパク質あるいは核酸をリガンドとして、受容体との結合様式を予測します。
- 外部プログラムの利用
活性部位への化合物の配置に、外部のドッキングプログラム*(FlexX、GOLD、Surflex-Dock) を利用できます。
また弊社では、MOE用ドッキングプログラムとして、AS_Dock、Ph4Dock、ASEDock (Goto J. et al, 2008)を開発しています。これらのドッキングプログラムは、上記の方法と異なる方法を利用してリガンドの結合様式を予測しており、より精度の高いモデル化と計算の高速化を実現しています。
結合自由エネルギーの推算
AMBER*の熱力学的積分法のインターフェースにより、相対的なリガンド-受容体間の結合自由エネルギーΔΔGを簡単に計算できます。
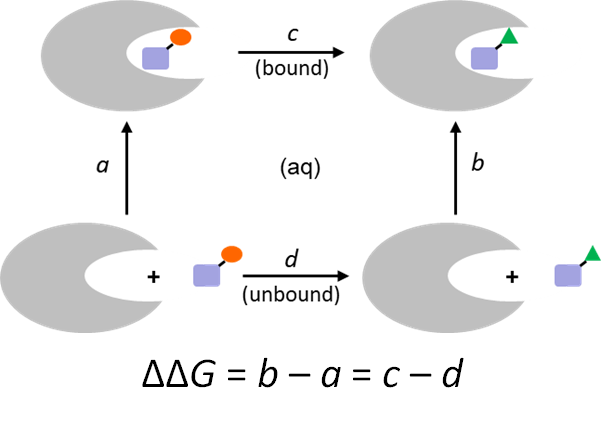
同じ結合モードを持つ二つのリガンドの相対的な結合自由エネルギーを計算
リガンド結合部位探索
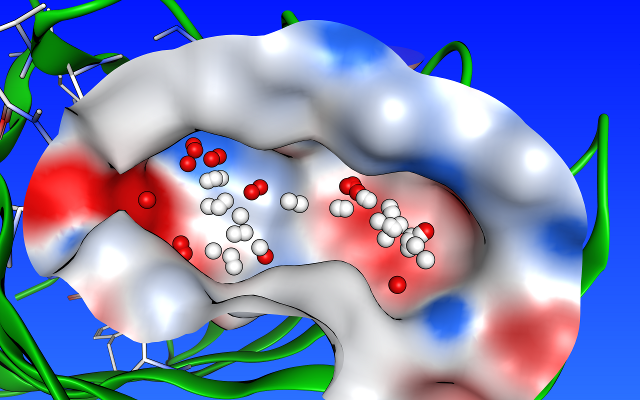 タンパク質の活性部位候補となりうるポケット構造を自動的に検出することができます。各ポケットに対して、アルファ球と呼ばれる疎水性(白色)または親水性(赤色)の小球を配置し、各ポケット内における疎水性・親水性領域の分布をグラフィカルに表示します。
タンパク質の活性部位候補となりうるポケット構造を自動的に検出することができます。各ポケットに対して、アルファ球と呼ばれる疎水性(白色)または親水性(赤色)の小球を配置し、各ポケット内における疎水性・親水性領域の分布をグラフィカルに表示します。この機能は、下記のドッキングシミュレーションのためのリガンド結合部位の選定だけでなく、既存のリガンドの修飾や新規リガンドの設計、ファーマコフォアモデルの作成にも利用することができます。
Protein Ligand Interaction Fingerprints (PLIF)
タンパク質-リガンド間の相互作用の種類と強さをフィンガープリントで表現し、複数の結合状態を統計的に解析します。ドッキング結果や複数の複合体構造に含まれる相互作用を解析することで、活性/不活性に関連する相互作用の検出や、活性/不活性を分類する相互作用組み合わせルールの抽出、ルールに適合する活性ポーズに共通するファーマコフォアの検出などが行えます。

リガンド結合部位2次元図
タンパク質-リガンド複合体からリガンド結合部位の2次元図を表示します。周辺残基の位置関係や相互作用、溶媒露出度が直感的に分かるように表示されます。水素結合、イオン結合、π-π相互作用、CH-π相互作用、カチオン-π相互作用、ハロゲン結合、配位結合などの相互作用を、それぞれ色分けして表示します。
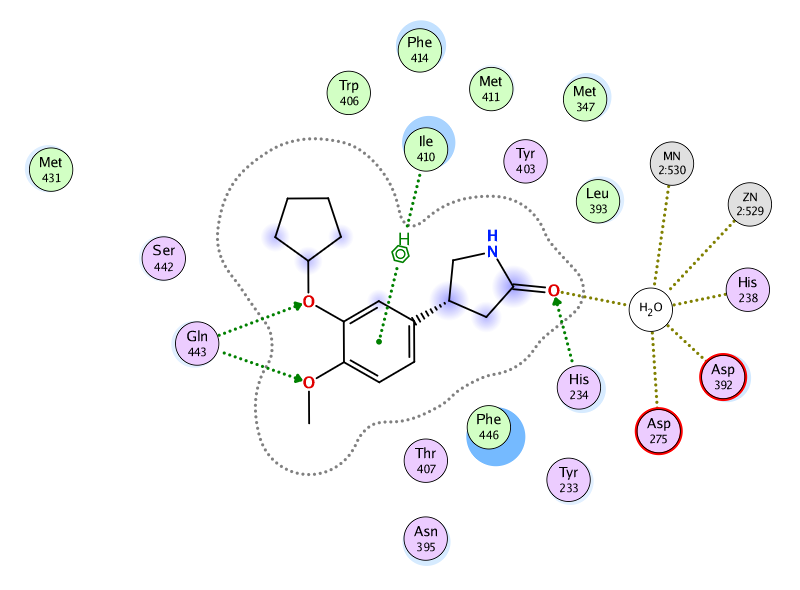
PDB ID: 1RO6のリガンド結合部位の2次元図
結合部位での分子設計
受容体に結合したリガンド構造を、より良好な活性や物性をもつように対話的に編集できます。結合したリガンド構造のうち、置換基を付加できる空間的な余裕のある箇所は分子グラフィックス上で緑色のベクトルとして表示されます。リガンド構造の分子量やTPSA、logP、logD、logSなどの物性、リガンドのひずみエネルギー、受容体との親和性、毒性に関わる部分構造の有無などの値は常に表示され、リガンドの改変と同時に更新されます。 ライブラリーからの置換基の自動探索や、サードパーティの2Dスケッチャーを用いた分子構造の改変も可能です。
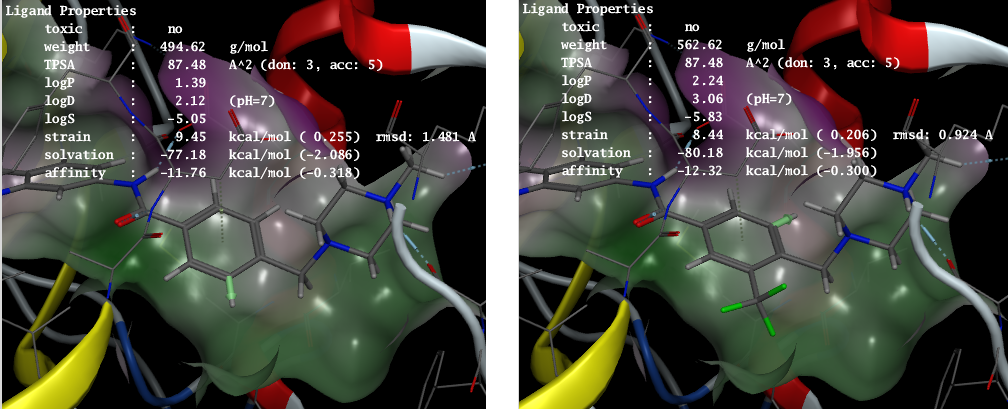
Bcr-Ablキナーゼimatinib複合体(PDB ID: 1IEP)。左図のベクトル(ベンゼン環の下に付加された緑の矢印)で示唆されたポケットを充填する位置へトリフルオロメチル基を付加(右図)することにより親和性が向上。
3D-RISM∗法による溶媒解析
∗Kovalenko, A.; Hirata, F. Self–consistent description of a metal–water interface by the Kohn–Sham density functional theory and the three–dimensional reference interaction site model; J. Chem Phys. 1999, 110, 10095-10112.
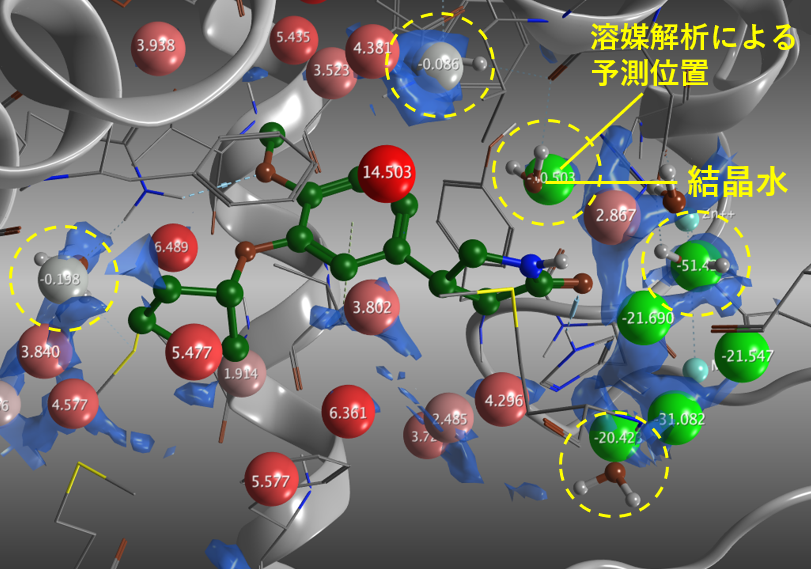
PDE4Bと結合したロリプラム複合体(PDB ID: 1RO6)の解析結果。水色の領域は水の確率密度がバルクの4倍の等値面。緑(ΔG負:安定)~赤(ΔG正:不安定)の球は予測された水分子の酸素原子の位置(Water Site)。いくつかの球はBall and Stick表示の結晶水近くに位置している。
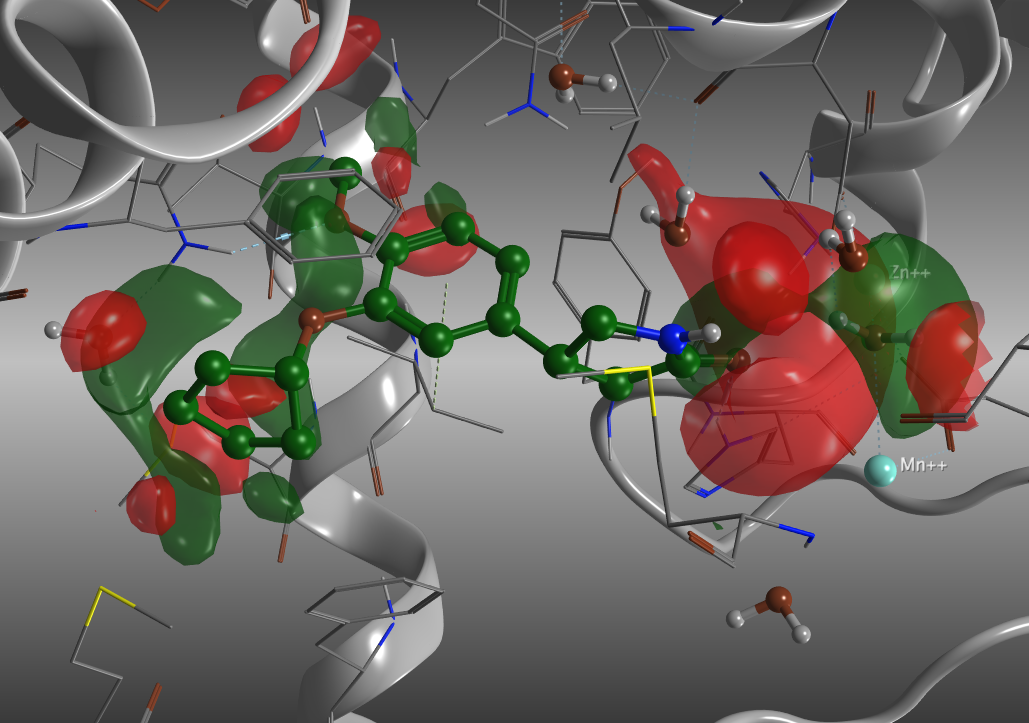
溶媒和エネルギーの等値面の表示。緑は負の値の領域で、リガンドの結合による脱溶媒で安定化される領域。赤は正の値の領域、リガンドの結合による脱溶媒により不安定になる領域。
分子表面解析
分子の表面形状として、溶媒接触表面とInteraction表面の2種類を描画することができます。溶媒接触表面はプローブ球の接触可能な表面を描画します。 Interaction表面は、受容体原子とプローブ球とのvan der Waals相互作用エネルギーがほぼ 0 kcal/molとなる面を描画します。それぞれの表面は、水素結合性、静電ポテンシャル、凹凸などを指標にした色分けにより、分子表面の特性を視覚的に解析できます。

分子表面付近の特性解析
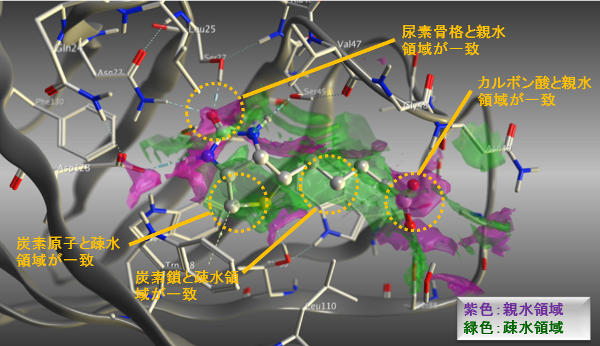
表面に対する相互作用相手の原子として好まれる特性領域をグラフィカルに表示します。領域は静電ポテンシャルや、PDBデータに対する統計解析、相互作用エネルギーなどを基に予測されます。複合体構造における相互作用の安定性に寄与している部分構造の予測や、分子設計の改変に適した箇所を検出できます。
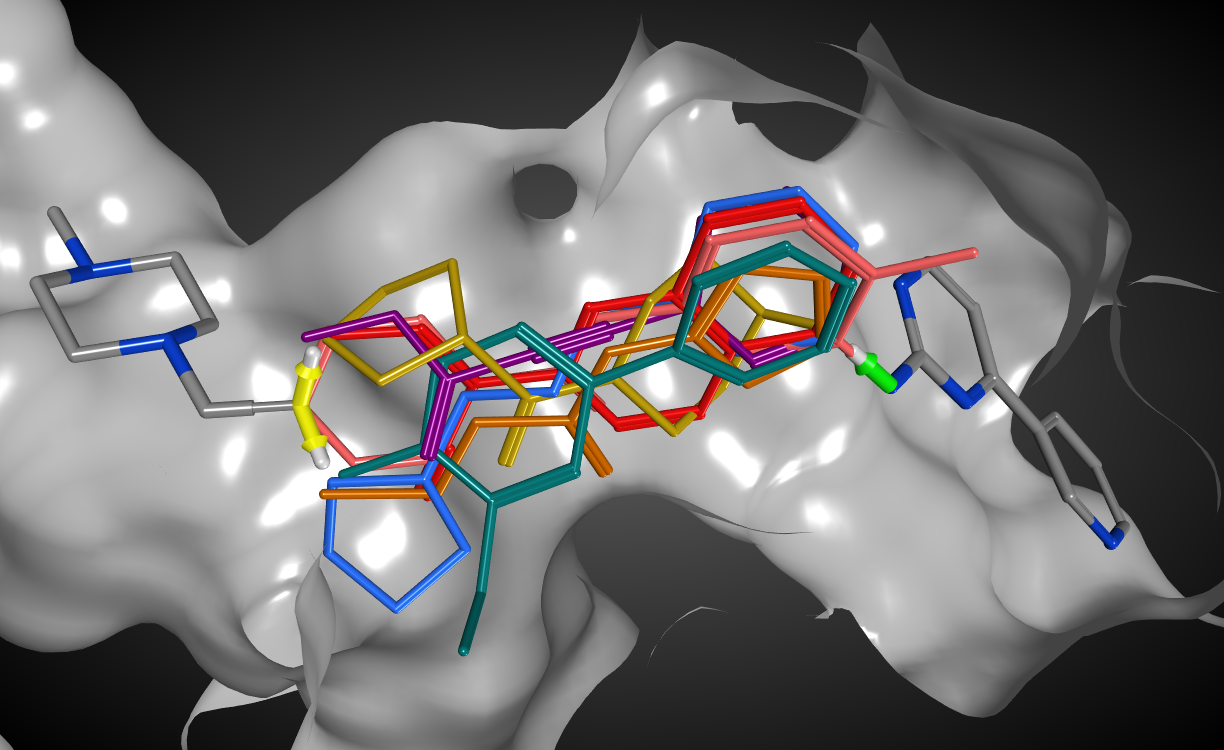
母核構造置換/フラグメント付加・連結
結合状態が既知のリガンドやドッキング構造に対して、母核構造の置き換えや官能基の付加、複数フラグメントの結合を行うことで、新規リガンド候補を提案します。MOEに付属する80万件以上のフラグメントデータベースもしくはin-houseフラグメントデータを結合して、受容体ポケット空間を考慮した衝突のない構造を提案します。
フラグメントの結合条件として、結合の方向と組み合わせを2種類のベクトルで定義できます。これにより結合距離や結合角の妥当な構造を探索でき、様々な結合の組み合わせを同時に計算できます。また、環化や環縮合を伴う結合条件も指定できます。
得られた構造は分子記述子やQSARモデル式、ファーマコフォア条件によって絞り込むことができます。さらに、合成可能性スコアや、ドッキング同様の構造最適化計算と親和性スコアによる順位付けから、実験に使用する構造の優先度を決定できます。
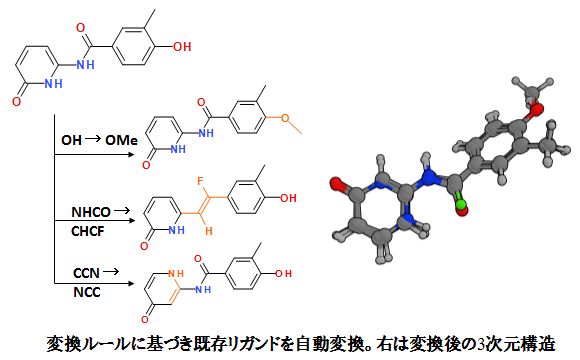
構造変換ルールによる新規分子構造への展開
既存リガンドを、構造変換ルールに基づいて分子構造を自動的に改変します。メディシナルケミストの知見を反映させた生物活性を保ちうる175種の部分構造変換ルールが標準で搭載されており、ルールはRXNファイルを利用して追加・編集が可能です。
新規リガンドは元のリガンド配置を考慮して再配置されます。母核構造置換同様、受容体、ファーマコフォアなどを考慮した変換が可能です。
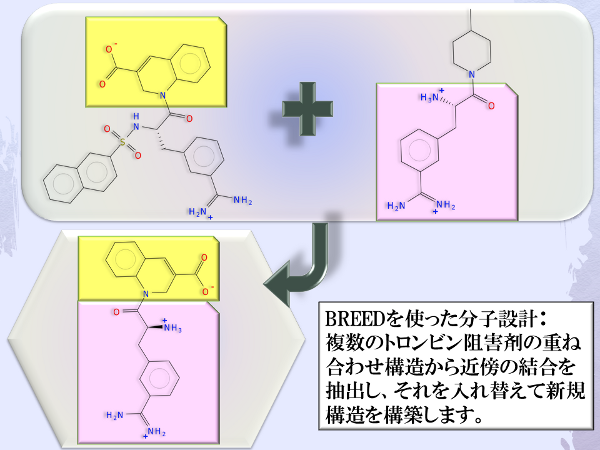
置換基の交換による新規リガンド設計 (BREED)
フラグメントデータベース
付属の76万件のフラグメント構造データベースや81万件のChEMBLフラグメントデータベースが提供されています。これらをフラグメントベースの分子設計に利用することができます。
MOEプロジェクトデータベースの構築
SBDD研究には、PDBの構造以外にも多種多様な情報が用いられます。MOE Projectデータベースには、標的タンパク質の活性部位での重ね合わせ構造や、リガンド構造、重要残基の構造アノテーション、アラインメント情報、リガンドの物性値、電子密度、実験値などのSBDDに活用できる情報を一つのデータベースに収録できます。創薬やエピジェネティックターゲットに利用できるMOE Projectデータベースが標準で搭載されています。MOE Projectデータベースは、ユーザー独自の定義で作成することができます。
作成されたMOE Projectデータベースから、キーワードや種、配列相同性、リガンドの類似度、ポケットの類似度、構造アノテーション(例 キナーゼのDFG in, out)等で柔軟な検索を行う為のインターフェースが搭載されています。ヒットした構造の可視化、統計情報、PLIF解析が可能です。
その他のデータコンテンツについてはこちらをご参照ください

相互作用解析に必要な前処理を一括実行(QuickPrep)
受容体-リガンド複合体の立体構造から相互作用解析やリガンドデザインを行うためには複数のステップからなる前処理が必要です。煩雑になりやすいこれらの前処理は統合されており、立体構造の取得からスムースに相互作用解析・ドッキング計算を行うことができます。取得した立体構造に対して、Protonate3Dによる高速な水素原子付加状態の最適化、相互作用に関与しない溶媒分子の除去、そして受容体構造を保持したまま相互作用部位周辺のひずみエネルギーを除く構造最適化といった前処理を一括して行います。処理内容の編集や保存も可能です。
- PSILO
- タンパク質立体構造情報データベースシステム
- Daylight
- 情報化学システム構築ツール
- Chemotargets CLARITY
- ターゲット・作用機序予測プラットフォーム