概要
ReaxFF
 反応分子動力学計算プログラム
反応分子動力学計算プログラム
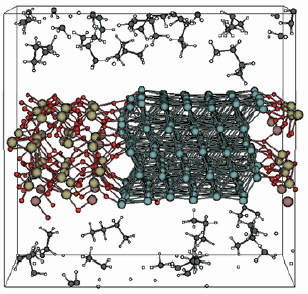 ReaxFF は、ペンシルベニア州立大学のvan Duin 教授らによって開発された反応分子動力学計算プログラムです。ReaxFF は、既存の古典分子動力学プログラムとは異なり、結合の生成と開裂を記述することができる反応力場 (Reactive Force Field)1 を搭載しています。反応力場のパラメータは量子化学計算のデータから決められているために化学反応の高精度な記述を可能にする一方で、その動力学計算は通常の古典分子動力学シミュレーションに匹敵する計算速度を保持しており、触媒反応や燃焼反応など、材料科学分野における分子動力学シミュレーションの適用範囲を大幅に広げています。
ReaxFF は、ペンシルベニア州立大学のvan Duin 教授らによって開発された反応分子動力学計算プログラムです。ReaxFF は、既存の古典分子動力学プログラムとは異なり、結合の生成と開裂を記述することができる反応力場 (Reactive Force Field)1 を搭載しています。反応力場のパラメータは量子化学計算のデータから決められているために化学反応の高精度な記述を可能にする一方で、その動力学計算は通常の古典分子動力学シミュレーションに匹敵する計算速度を保持しており、触媒反応や燃焼反応など、材料科学分野における分子動力学シミュレーションの適用範囲を大幅に広げています。SCM 社ではReaxFF コードに対して並列化を含む各種最適化を行っています。また、メモリ使用量などの最適化により10万原子程度を含む3 次元周期系の計算が通常のデスクトップPC上で対応可能です。また、ReaxFF にはグラフィカルユーザインターフェース(GUI)が用意されており、バルクモデルの構築、分子動力学の計算設定、ジョブ投入、計算結果の可視化に対応しています。
1) A.C.T. van Duin, S. Dasgupta, F. Lorant, and W.A. Goddard III, J. Phys. Chem. A 105, 9396 (2001).
ReaxFFの主な機能
- 反応力場による構造最適化と分子動力学計算 (アンサンブルはNVE, NVT, NPT に対応)
- 3 次元バルクモデルの構築(Packmol を使用した溶媒分子のランダム配置に対応)
- シミュレーティッドアニーリングおよび異なる温度領域の設定
- 分子動力学計算で得られた座標履歴のアニメーション表示と各種物理量の時間発展のグラフ表示
- PyMDとのインターフェース(メタダイナミスクやアンブレラサンプリングなどの自由エネルギー計算に対応)
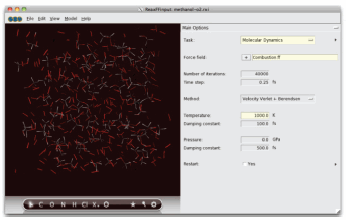
[ReaxFF-GUI によるバルクモデル構築と計算設定]
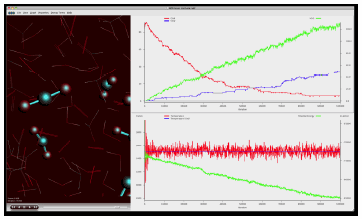
[反応中に生成した水分子の総数の時間発展のグラフ表示]