


MOE - タンパク質、核酸モデリング
tab:{
title:{概要}
MOEでは、タンパク質、核酸モデリングを支援する多くの機能が搭載されています。タンパク質モデリングにおいては、アミノ酸配列から立体構造を予測するホモロジーモデリング、融合タンパク質の構築や電子密度に適合したループ構造の補完などに活用できるループ/リンカーモデリング、単量体から複合体を予測するタンパク質-タンパク質ドッキングなどがあります。核酸モデリングにおいては、A/B/C型などのらせん構造を配列情報から構築する機能や既存の核酸を非天然塩基を導入する機能などがあります。さらに、モデリングした構造について、主鎖二面角のプロットなどの構造品質評価、相互作用解析、分子表面の可視化などを行えます。
title:{タンパク質立体構造予測}
MOEでは、ホモロジーモデリングの機能により、アミノ酸配列からタンパク質立体構造を予測できます。ホモロジーモデリングは、タンパク質のアミノ酸配列の相同性が高ければ立体構造の類似性が高いという性質を利用します。立体構造が既知であるファミリータンパク質の立体構造を鋳型として、その鋳型に標的タンパク質のアミノ酸配列を当てはめるようにしてタンパク質の立体構造モデルを構築します。鋳型タンパク質を参照できない部分的な主鎖構造は、セグメントマッチ法により他の立体構造データを割り当て、側鎖構造は最適なパッキングとなるものをライブラリーから選択します。MOEでは、(1)相同タンパク質の検索、(2)多重配列アラインメント、(3)構造保持領域/相互作用解析、(4)タンパク質立体構造構築、(5)タンパク質構造解析というステップで、類縁構造の検出から相互作用を考慮したアラインメントの補正、得られたモデルの評価を行えます。
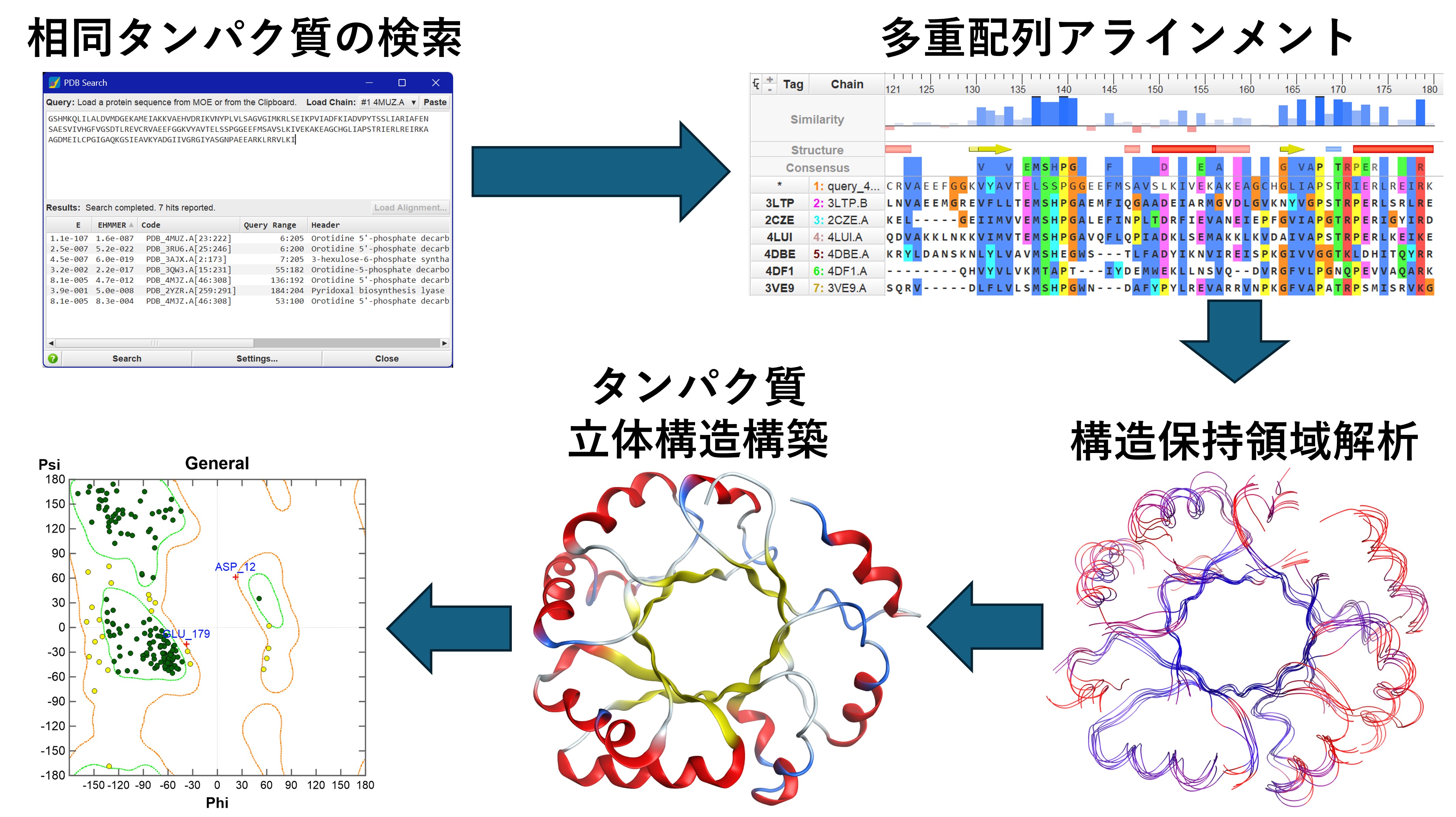
MOEでは、単量体だけでなく、複数のタンパク質をテンプレートとすることで、多量体モデルも構築できます。また、タンパク質立体構造構築の際に、リガンドや水分子、金属イオンなどのヘテロ原子を考慮できます。複数のタンパク質をテンプレートとしたモデリングも可能です。
title:{DNA/RNAの構築}
MOEでは塩基を順番に選択、あるいは、塩基配列を入力するだけで目的の核酸を構築できます。A、B、C、Z型などの二重らせん、あるいは、一重らせんの構造を構築できます。一重らせん構造からの相補的な二重らせん構造への補完も可能です。DNAからRNAまたはその逆の変換、指定したらせん構造への変換が可能です。天然塩基への変異体の構築や、特殊塩基を用いた変異体も構築できます。特殊塩基はユーザー定義の塩基を追加できます。
MOE核酸モデリングパンフレット
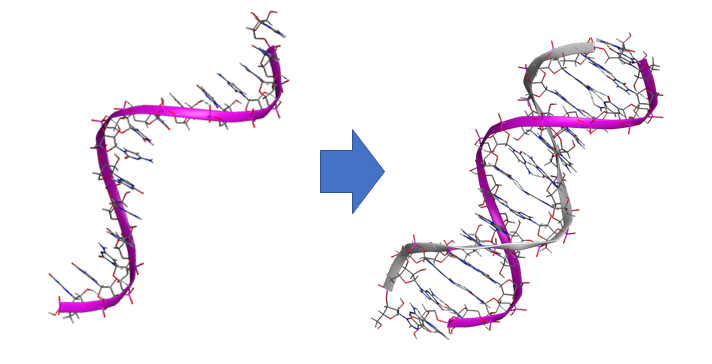
一重らせん構造から二重らせん構造への相補的配列と構造の自動補完。アンチセンス核酸医薬設計を支援。
title:{相互作用と分子表面の可視化}
相互作用の可視化
MOEのタンパク質の相互作用解析機能では、水素結合、イオン相互作用、vdW相互作用、共有結合、CH…O相互作用、π…H相互作用、ハロゲン結合、配位結合、アレーン接触を検出できます。分子内相互作用やタンパク質間相互作用(Protein-Protein Interaction)を解析できます。残基番号を軸の値に利用した2次元グラフを表示できます。データ点を選択すると、対応する3次元構造を選択・可視化が容易にできます。

上皮成長因子受容体で保存された分子内相互作用の2次元グラフ(左)、選択データ点の3次元構造(右)
分子表面の可視化
疎水性、正電荷、負電荷の傾向が強いタンパク質表面の一部(表面パッチ)を可視化できます。表面パッチは、タンパク質の凝集やタンパク質間相互作用、溶解度などに影響します。凝集を防ぎ溶解度を向上するための残基の特定や、PPI阻害剤のターゲットの探索に活用できます。表面パッチの二次元への投影図は、野生型と変異体との比較などに利用できます。
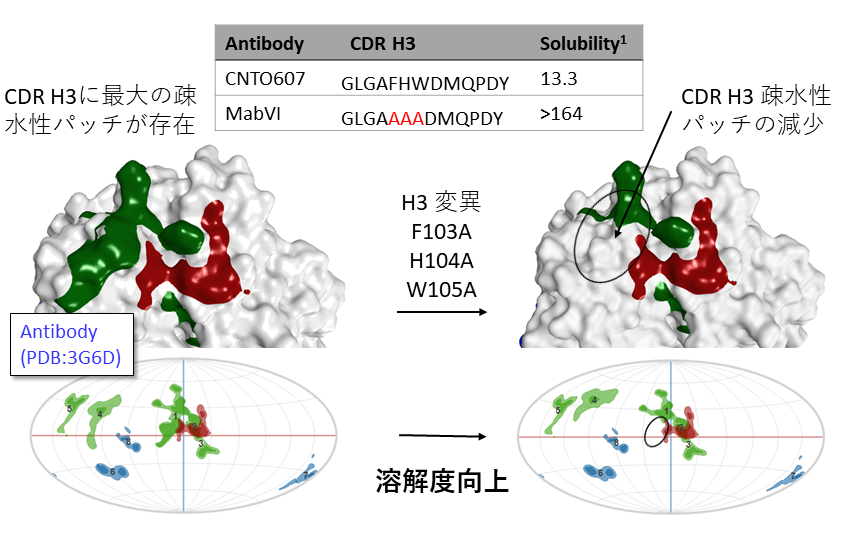
抗体CNTO607(左図)は高濃度で凝集。CDR H3にある疎水性パッチが多量体形成の原因となりうる。疎水性パッチ領域にアラニンを導入した変異体(右図)は疎水性パッチが減少し溶解度の向上が見られた。
Wu, S.-J.; et al. Protein. Eng. Des. Sel. 2010, 23 (8), 643–651.
title:{変異体と側鎖配座の探索}
ターゲットとなるアミノ酸残基や塩基を指定するだけで、変異体の配座候補を得られます。事前に用意された側鎖配座データベースから、立体障害のない候補構造を提案します。それぞれの候補はファンデルワールスエネルギー、水素結合エネルギーなどによってランク付けされます。特殊アミノ酸、特殊塩基への変異も可能です。ユーザーオリジナルの特殊アミノ酸、特殊塩基を登録し変異体構築に使用できます。
title:{分子動力学シミュレーション}
分子動力学(MD)シミュレーションでは、分子を熱運動させて、位置や速度、エネルギーなどについて動的経過を解析します。MOEでは、MDシミュレーションの一連の作業フローを支援します。初期構造の準備においては、タンパク質構造の前処理、水素結合ネットワークを考慮した水素付加状態の最適化、周期境界条件の設定や溶媒分子/カウンターイオンの配置ができます。構造最適化計算では、MOE独自の分子力場であるAmber:EHTが使用でき、幅広い分子系について精度よくエネルギーを評価できます。計算エンジンとしてNAMD*やAMBER*が利用でき、マルチコア、クラスター、GPUを利用した計算設定にも 対応しています。受容体と複数のリガンドの構造群から、 計算に必要なインプット/スクリプトファイルを自動的に作成します。MDシミュレーションの結果をMOEに取り込んで、RMSD、RMSF、原子間のジオメトリー解析などのさまざまな解析が行えます。
*別途入手が必要です。

HIVプロテアーゼと阻害剤の複合体に周期境界条件を設定した分子動力学計算の初期構造
title:{ループ/リンカーモデリング}
MOEには、ループ/リンカー探索とサンプリングが行えるループ/リンカーモデリング機能があります。この機能では、タンパク質のループ構造を対話的に構築できます。ループ構造の構築手法として、PDBデータ内から検索する経験的探索と、新規のループ構造を構築するde novo探索の二種類が提供されています。前者は高速に構築でき、鋳型との配列相同性を確認でき、後者は全く新規のループを構築でき、前者でヒットが得られない場合に有用です。得られたループの順位付けには、溶媒和を考慮した相互作用エネルギーや、リガンドとの親和性、電子密度マップとの適合性に基づくスコアが利用できます。リンカーモデリングでは、2つのタンパク質を融合するためのリンカー候補を探索します。この機能は、SBDD分野におけるタンパク質のループ構造のサンプリング、CABD (Computer-Aided Biologics Design)分野における融合タンパク質の構築、結晶解析分野における電子密度によるループ構造の補完など幅広い分野に活用できます。
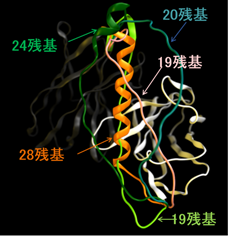
VLとVHと一本鎖抗体の構築例(PDB ID:1VFB)。
また、タンパク質の任意の部分構造をほかの構造より移植するループグラフティングもできます。

c-Ablキナーゼの活性化ループのループグラフティングのプレビュー表示(橙:移植前のループ、緑:移植後のループ、青:移植先の隣接残基、水色:移植元の隣接残基)
title:{タンパク質-タンパク質ドッキング}
タンパク質-タンパク質あるいは核酸-タンパク質間のドッキングシミュレーションは、単体のタンパク質、核酸の構造から、それらが形成する複合体の構造を予測します。得られた複合体の構造はタンパク質の重要部位の特定(エピトープマッピング)、タンパク質間の相互作用の解析・設計に利用できます。計算はアミノ酸や核酸の複数の構成原子を一つのビーズに置き換えた粗視化モデルとグリッドを用いて高速化しています。探索範囲を抗体のCDRや酵素活性部位などに限定して計算を行えます。
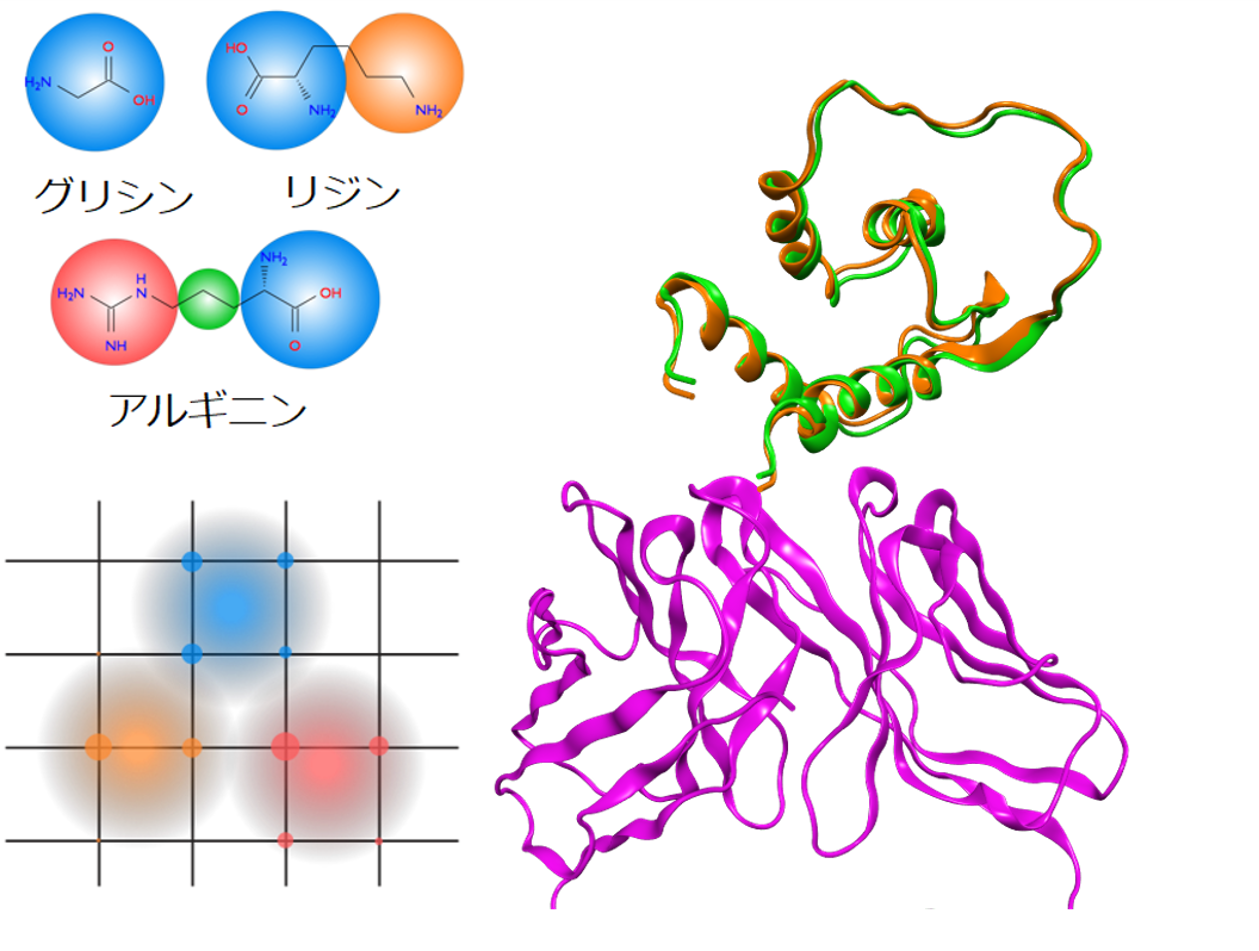
アミノ酸残基の粗視化イメージ(左上)、リガンドのガウス関数で重み付けされた密度のイメージ(左下)、抗体(右下、紫)とインターロイキン13(右上、実験結果:橙、計算結果:緑)のドッキング構造(PDB ID: 3G6D、RMSD: 0.8 Å)
}:tab