

モジュール
 力場データベース TEAM_Family、AMBER_Family
力場データベース TEAM_Family、AMBER_Family
 一般的な力場では、原子タイプ割付ルールと力場パラメータは一組のみ取り扱われますが、力場データベース(以下力場DB)では、異なる原子タイプ割付ルールと力場パラメータをもつ力場を、複数同時に整合性を保ちながら統合し、利用する機能を備えています。この機能により、力場タイプが同一であれば、一部をユーザオリジナルの力場、他の部分をDFFの提供する力場というようにして、別々に開発された力場パラメータを矛盾なく1つのパラメータセットとして利用することが可能です。また、原子タイプ割付ルールを簡単に編集する機能を備えているので、デフォルトより詳細なユーザ独自の割付ルールを作成することも可能です*4。これらにより、力場パラメータ代用の問題と原子タイプの重複による矛盾を解決しながら、力場パラメータの精度と拡張性を同時に向上させています。
一般的な力場では、原子タイプ割付ルールと力場パラメータは一組のみ取り扱われますが、力場データベース(以下力場DB)では、異なる原子タイプ割付ルールと力場パラメータをもつ力場を、複数同時に整合性を保ちながら統合し、利用する機能を備えています。この機能により、力場タイプが同一であれば、一部をユーザオリジナルの力場、他の部分をDFFの提供する力場というようにして、別々に開発された力場パラメータを矛盾なく1つのパラメータセットとして利用することが可能です。また、原子タイプ割付ルールを簡単に編集する機能を備えているので、デフォルトより詳細なユーザ独自の割付ルールを作成することも可能です*4。これらにより、力場パラメータ代用の問題と原子タイプの重複による矛盾を解決しながら、力場パラメータの精度と拡張性を同時に向上させています。製品付属する力場DBには、TEAM_Family、AMBER_Familyの2つが用意されており、用途に応じていずれかの力場DBを選択していただくことになります。材料設計向け力場DB、TEAM_Familyは関数形にAeon社オリジナルのTEAMを採用し、有機低分子、合成高分子、イオン液体、ゼオライト用のパラメータが登録されています。ライフサイエンス向け力場DB、AMBER_Familyは関数形にAMBERを採用し、TEAM_Family同様、有機低分子、合成高分子、イオン液体のパラメータが登録されており、さらに公開されているAMBER95 のパラメータが登録されています。
※4. Professionalのみの機能です。
- 力場データベースにおけるデータフィッティングの信頼性
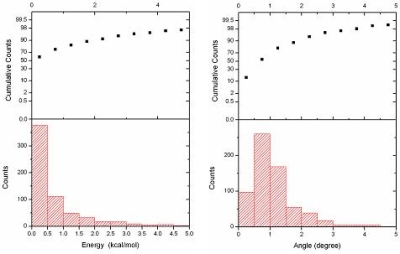
- 量子化学計算とのコンフォメーションのエネルギー誤差(左)と最適化構造の結合角の誤差(右)の分布のグラフとその積算率。コンフォメーションのエネルギー誤差が3kcal/mol以下のものが95%、結合角の誤差は3度以下のものは約98%と、サンプリングに利用した量子化学計算と同程度の精度が望めます。
- 力場パラメータ代用の問題
- 力場の精度は、力場パラメータの数値の精度だけでなく、分子中の多様な原子の環境を解釈して如何に妥当な力場パラメータを原子にアサインできるかに大きく依存します。一般の力場の二面角(A-B-C-D)の力場パラメータは、連なる4原子の2番目(B)と3番目(C)の原子タイプによりアサインされます。このような場合、1番目(A)と4番目(D)の原子がどのような原子タイプであっても、全く同じ力場パラメータを利用してしまうため、二面角の回転障壁の形が全く異なってしまうことが多々あります。このようなことを力場パラメータの代用の問題といいます。
GUI機能
分子構造の構築、可視化、さまざまな分子フォーマットの読み込みを可能にします。さらに、力場の開発だけでなく、一般的な分子モデリングのツールとしても利用できます。低分子液体モデリング機能、自動電荷グループアサイン機能等が搭載されています。
シミュレーション機能
DFFの力場データベースで得られた力場パラメータや開発された力場パラメータを、DFFの分子シミュレーションエンジンやサードパーティの分子シミュレーションエンジンで利用するための機能です。DFFの分子シミュレーションエンジンは力場パラメータの検証やVDWパラメータ作成に必要な分子動力学計算データを用意するのに利用されます。サードパーティの分子シミュレーションエンジンに対しては座標データ、トポロジーデータ、原子タイプデータ等、計算に必要なデータの作成を行います。
力場パラメータ開発機能
 量子化学計算の結果から力場パラメータの作成するための機能です。非常に複雑なポテンシャル面に、力場のポテンシャル関数を効率よくフィットするパラメータ決定手法を備えており、量子化学計算のデータから信頼性の高い力場パラメータを高速に開発できます*5。
量子化学計算の結果から力場パラメータの作成するための機能です。非常に複雑なポテンシャル面に、力場のポテンシャル関数を効率よくフィットするパラメータ決定手法を備えており、量子化学計算のデータから信頼性の高い力場パラメータを高速に開発できます*5。本機能には、力場パラメータを作成するのに必要な部分構造(フラグメント)に対して、量子化学計算データを作成、読み込みをするためのインターフェースが含まれています*6。力場パラメータ作成には、フラグメントに対して、最安定構造の探索、基準振動解析、パラメータフィッティングに利用する配座の一次微分の計算を行います。本来、これら手順は、全てユーザが一つ一つコマンドを実行して行う手間のかかる作業ですが、1つのボタンをクリックするだけで自動的に行うこともできます*7。
凝集系のシミュレーションで高い精度を要求されるvan der Waals (VDW)パラメータについては、系の密度と蒸発熱の実験値から自動的にVDWパラメータをフィッティングする独自の手法が搭載されています*8。
さらに、予め用意されている力場のポテンシャル関数でよく利用されるさまざまな項のテンプレートを自由に組み合わせることで、ユーザオリジナルの関数による力場パラメータ開発も可能です。これにより、ライフサイエンスからマテリアルサイエンスまで、さまざまな分子シミュレーションに対応できる力場を開発することが可能です。
※5. Sato, F.; Hojo, S.; Sun, H. J. Phys. Chem. A, 2003, 107, 248-257
※6. Gaussianに対応。量子化学計算ソフトウェアGaussianが別途必要です。
※7. GaussianがDFFの起動できるPCにインストールされている必要があります。
※8. Sun, H. Fluid Phase Equilibria, 2004, 217, 59-76.
※6. Gaussianに対応。量子化学計算ソフトウェアGaussianが別途必要です。
※7. GaussianがDFFの起動できるPCにインストールされている必要があります。
※8. Sun, H. Fluid Phase Equilibria, 2004, 217, 59-76.
- SciMAPS
- 材料設計支援プラットフォーム