
機能詳細
モンテカルロ計算プログラム MedeA-GIBBS
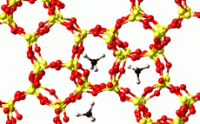 GIBBS は、気体・液体の熱力学物性をモンテカルロ法により推算するプログラムです。
GIBBS は、気体・液体の熱力学物性をモンテカルロ法により推算するプログラムです。
低分子の液液・気液平衡や、低分子・長鎖のナノポーラス物質への吸着現象など、様々な系に適用することができます。
 機能
機能
- アンサンブル
-
- カノニカルアンサンブル(NVT)
- 定圧アンサンブル(NPT)
- グランドカノニカルアンサンブル(μVT)
- ギブスアンサンブル(定積、定圧)
- ポテンシャル
-
- OPLS-UA
- TraPPE
- Anisotropic United Atom (AUA)
- 相互作用エネルギーの評価
-
- 分散・反発相互作用(Lennard-Jones、Buckingham)
- 交差項には4 つの混合ルールが適用可(Lorentz-Berthelot、Kong、Waldman-Hagler、geometric)
- 静電相互作用(Cutoff、Reaction Field、Ewald)
- 遠距離相互作用のカットオフに対する補正
- 分子自由度
-
- 剛体モデル
- フレキシブルモデル
- 半剛体モデル
- 分子移動手法
-
- Configurational Biased モンテカルロ法による長鎖の取り扱い
- リザーバーバイアス法によるフレキシブルモデルの取り扱い
物性
- 単一層における熱力学物性
-
- 圧力、体積、密度、熱容量、熱力学関数等の温度変化
- 化学ポテンシャル
- Joule-Thomson 係数
- 相平衡
-
- 気液平衡(VLE)、液液平衡(LLE)
- 気液液平衡(VLLE)
- 蒸気圧曲線
- ナノポーラス物質への分子の吸着現象
-
- 吸着等温線
適用例
 faujasiteへの吸着による m-キシレンと p-キシレンの分離を検討した結果;
faujasiteへの吸着による m-キシレンと p-キシレンの分離を検討した結果;
気相中の p-キシレンのモル分率に対する固相中の p-キシレンのモル分率 (X-Y 図)を推算した結果は、実験結果をよく再現しています(*1)。
1. V. Lachet. Boutin, B. Tavitian, and A. H. Fuchs, Langmuir 15, 8678(1999).
分子動力学計算プログラム MedeA-LAMMPS
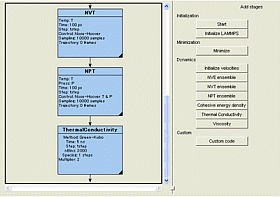 LAMMPS(*2)は、サンディア国立研究所(米国エネルギー省)によって開発されたプログラムで、世界中の様々な研究分野で利用されています。フローチャート機能によるタスク制御で、平衡化計算を柔軟に設計でき、さらに各種物性推算ツールと組み合わせることで、熱伝導性や拡散等を評価できます。
LAMMPS(*2)は、サンディア国立研究所(米国エネルギー省)によって開発されたプログラムで、世界中の様々な研究分野で利用されています。フローチャート機能によるタスク制御で、平衡化計算を柔軟に設計でき、さらに各種物性推算ツールと組み合わせることで、熱伝導性や拡散等を評価できます。
2. S. Plimpton, J. Comp. Phys., 117, 1 (1995); http://lammps.sandia.gov/index/?taxonomy=product_details-content
機能
- アンサンブル
-
- ミクロカノニカルアンサンブル(NVE)
- カノニカルアンサンブル(NVT)
- 定圧アンサンブル(NPT)
- ポテンシャル
-
- 有機分子・高分子用
OPLS-AA、PCFF、COMPASS(Published only)、CFF91、CFF93、CVFF - 無機物・酸化物用
Inorganic、CVFF_AUG、BKS - 半導体用
Stillinger-Weber、Tersoff
- 有機分子・高分子用
- 相互作用エネルギーの評価
-
- 静電相互作用(Cutoff、PPPM、Ewald)
- 遠距離相互作用のカットオフに対する補正
物性
- 純物質・混合物における流体の物性
-
- 圧力、体積、密度、エネルギー
MedeA-LAMMPSと組み合わせ可能な物性推算ツール
- Thermal Conductivity & Viscosity 熱伝導性・粘性評価ツール
- LAMMPS-Diffusion 拡散係数評価ツール
- LAMMPS-CED 凝集エネルギー密度評価ツール
- MedeA-MT 弾性特性・熱力学物性評価ツール
EAM力場パラメータ LAMMPS-EAM
LAMMPS-EAMは、Embedded Atom Method(原子挿入法)による力場です。金属を扱うのに適した力場で、第一原理計算で扱うには負荷の大きい大規模な系を分子動力学計算で扱うことができます。Zhouらのパラメータ(*3)をサポートしており、以下の金属を扱うことができます。
Cu, Ag, Au, Ni, Pd, Pt, Al, Pb, Fe, Mo, Ta, W, Mg, Co, Ti, Zr

3. X. W. Zhou, R. A. Johnson, H.N.G. Wadley, Phys. Rev. B. 69, 144113 (2004)
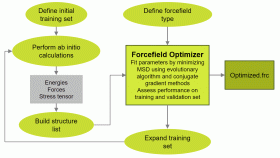
力場最適化ツール Forcefield Optimizer
FFO( Forcefield Optimizer )は、第一原理計算の結果を基に力場パラメータを最適化するツールです(*4)。新規の系に対して力場パラメータを最適化して、力場計算によるシミュレーションを可能にします。
トレーニングセットを作成するために第一原理計算プログラムMedeA-VASP を使用します。最適化された力場パラメータを検証するために分子動力学計算プログラムMedeA-LAMMPS を使用します。
機能
- 第一原理計算分子動力学計算の履歴から各ステップの情報を抽出:構造、エネルギー、力、応力
- どの力場パラメータを最適化するか選択可能
- 遺伝的アルゴリズムおよび最小二乗法によるパラメータの最適化
- 最適化された力場パラメータはファイルとして保存され、力場計算に利用可能
- 最適化可能な力場形式:Buckingham, EAM, PCFF, Tersoff
4. Forcefield Optimizerの方法論については次の文献を参照ください。
R. Asahi, C. M. Freeman, P. Saxe and E. Wimmer, Modelling Simul. Mater. Sci. Eng. 22, 075009 (2014).
M. Christensen, W. Wolf, C. Freeman, E. Wimmer, R. B. Adamson, L. Hallstadius, P. E. Cantonwine, E. V. Mader, Journal of Nuclear Materials 460, 82 (2015).
- SciMAPS
- 材料設計支援プラットフォーム