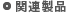シミュレーション
 シミュレーション機能 ~先進のプログラム群をサポート~
シミュレーション機能 ~先進のプログラム群をサポート~
さまざまなスケールの計算手法に対応したプラグインを分野ごとにまとめたパッケージとして提供します。パッケージには量子化学・物理計算(Quantum)、力場計算、メソスケール計算(Classical)、熱力学物性計算、構造活性相関(Engineering)の3つがあります。それぞれの計算分野で定評のある先進のシミュレーションプログラムに対応しています。
Classical(分子動力学計算、メソスケール計算)
分子動力学計算プログラム

また、粗視化モデルを構築して粗視化力場を割り付ければ、粗視化分子動力学計算も実行できます。
- □主な機能
- 極小化計算
- NVE、NVT、NPTの各種アンサンブル
- VDW相互作用計算 カットオフ法
- クーロン相互作用計算 カットオフ法、PPPM法、PME法、Ewald法
- マルチタイムスケール数値積分法(rRESPA法)
- DREIDING、CFF、AMBER、CHARMMの各力場に対応
- SDK、Martini、SAFT-γの各粗視化力場に対応
- □対応プログラム
- LAMMPS-Atomistic Plug-in
- GROMACS Plug-in
モンテカルロ計算プログラム
モンテカルロ法は、確率的手法を用いて系の状態を出現させることで、系を構成する粒子の分布を求める方法です。分子動力学計算のように時間の概念を考慮できませんが、分子動力学計算では取り扱うことの難しい液液、気液平衡などの相平衡状態やゼオライトへの吸着現象の研究に最適なプログラムです。また、SciMAPSがサポートするプログラムは、効率の良いコンフォメーションを発生する多数のアルゴリズムを用意し、さまざまな力場関数系に対応しています。
- □主な機能
- NVE、NVT、NPT、μVT、ギブスの各種アンサンブルに対応
- CBMC法による効率の良いサンプリング手法を搭載
- AMBER、CHARMM、DREIDINGの各力場に対応
- □対応プログラム
- Chameleon Plug-in
- Towhee Plug-in
- CASSANDRA Plug-in
散逸粒子動力学
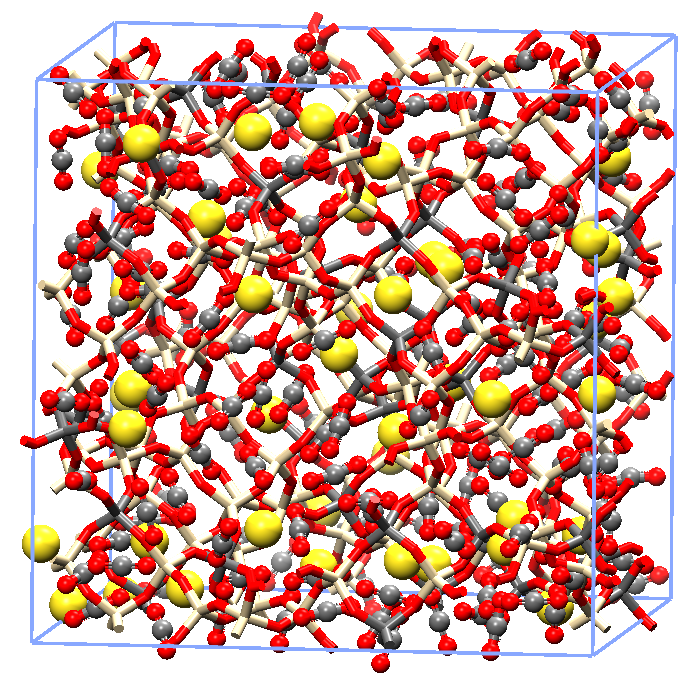
複数の原子、分子を1つの粗視化粒子(ビーズ)として表現し、ソフトマターをシミュレーションするプログラムです。散逸粒子動力学計算では、粒子間に働く力を散逸力、ランダム力、保存力の3つからなると仮定し、粒子間の重なりを許容するため、流体の挙動を表すのに適しています。混合液体の相分離構造、ミセルの形成、高分子バルクの多形予測、薬物の放出現象等、分子動力学では扱いが難しいさまざまな流体のシミュレーションを可能にします。
- □対応プログラム
- LAMMPS-DPD Plug-in
フラグメント間相互作用計算プログラム
フラグメント間相互作用計算プログラムはMolecular Silverware法による二元混合物の相互作用を計算します。高分子や液体の熱力学物性や散逸粒子動力学計算の相互作用パラメータの推算に利用します。
- □対応プログラム
- FHMixinig Plug-in
粗視化パラメータ計算プログラム
全原子での分子動力学計算で得られた構造や原子上に働く力の履歴データを基に、対応する粗視化モデルのビーズ間の相互作用を調べ、粗視化パラメータ計算手法に基づいて相互作用の値を求めます。最終的に得られるビーズ間相互作用の値はテーブル化した数値データポテンシャルとして出力します。計算手法としては、Iterative Boltzmann Inversion(IBI)法、Force Matching(FM)法の2つが用意されています。
- □対応プログラム
- VOTCA Plug-in
Quantum(量子力学計算)
第一原理バンド計算
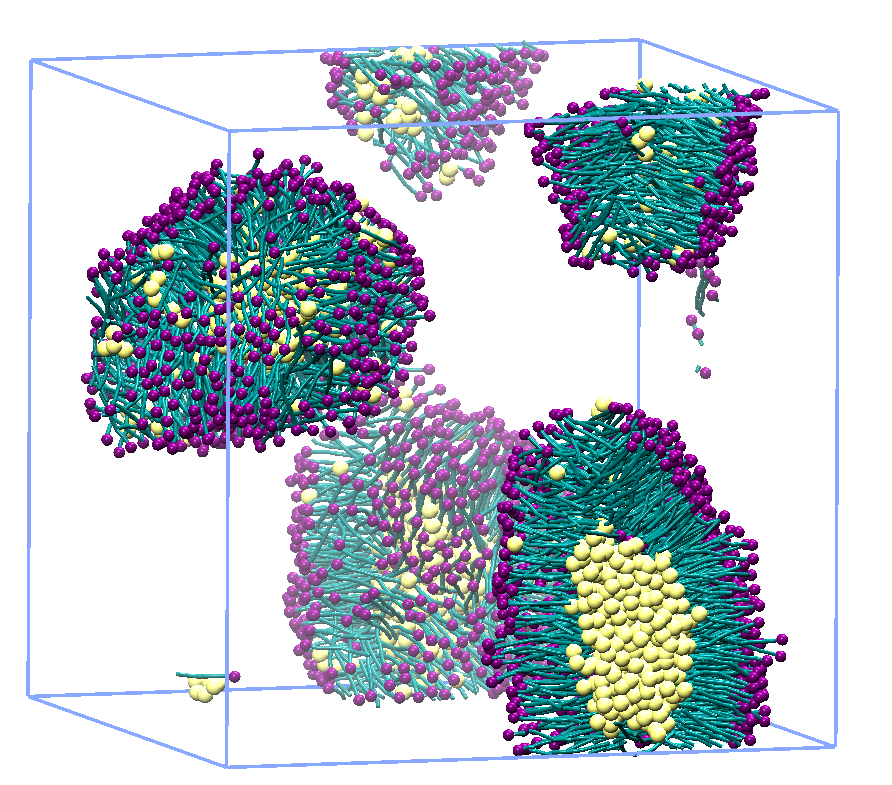
周期境界条件下で密度汎関数法を用いた平面波-擬ポテンシャル法電子状態計算のための量子力学計算プログラムです。時間依存DF T法やGW法、PAW法等、密度汎関数法のさまざまな技術により、バンド構造、状態密度、構造(結合長、結合角、プリミティブセルのサイズと形状等)、凝集エネルギー、誘電特性、振動特性等の計算が可能です。金属、セラミックス、半導体等の固体、表面、界面の研究に最適です。
- □対応プログラム
- Quantum Espresso Plug-in
- ABINIT Plug-in
- VASP Interface
量子化学計算プログラム
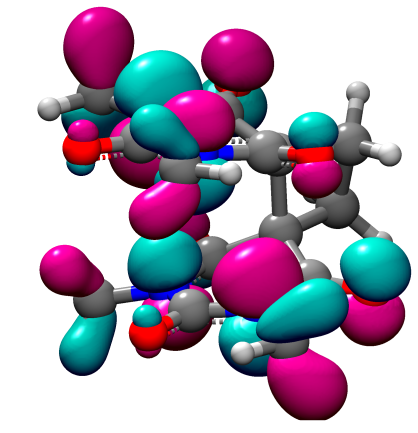
非周期境界条件下での量子力学計算プログラムです。HF、MP2、DF T、時間依存DF T、CC法等の計算手法に加え、さまざまな基底関数を選択できますので、ユーザの必要に応じた精度での計算が可能です。電子状態、分子構造(結合距離、結合角、二面角等)、各種スペクトル(赤外、ラマン、可視紫外)等の物性や反応経路におけるエネルギー(遷移状態、エネルギー障壁)の推算に最適です。
- □対応プログラム
- NWCHEM Plug-in
- TURBOMOLE Interface
半経験的分子軌道法プログラム
NDDO近似に基づき、実験値を再現するように決められた経験的なパラメータを用いることによって、計算コストをかけずに精度の高い計算を可能にしたプログラムです。非経験的分子軌道法では扱うことのできない大きな分子に対しても非常に高速に計算でき、低分子から数百原子程度の比較的大きな分子までの分子構造や物性の予測に適しています。
- □対応プログラム
- MOPAC Interface
- MNDO Plug-in
遷移状態探索プログラム
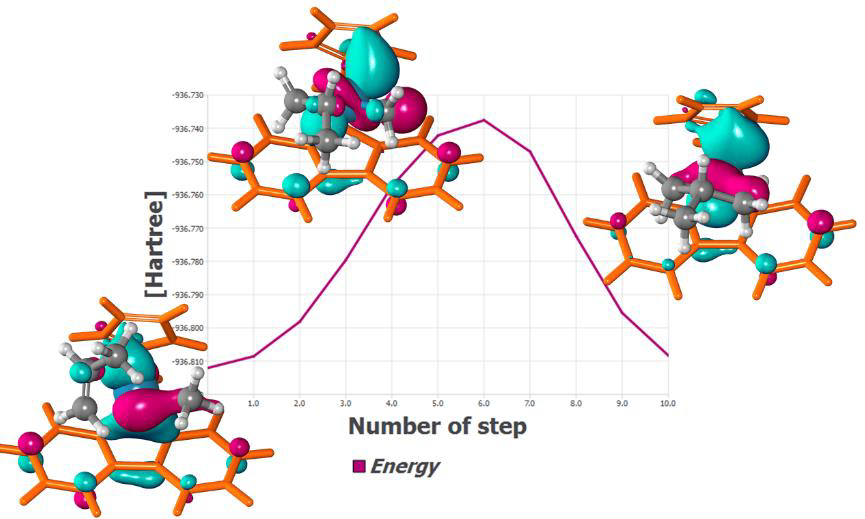
量子化学計算で得られるエネルギー表面上の遷移状態を探索するツールです。各量子力学計算プログラムで計算されたエネルギーおよびその一次微分の情報を使って、LST(Linear Synchronous Transit)法、QST(Quadratic Synchronous Transit)法およびNEB(Nudged Elastic Band)法が利用できます。
- □対応プログラム
- TRANSITION STATE LOCATOR
EngineeringE(熱力学物性計算、情報科学)
熱力学物性推算プログラム
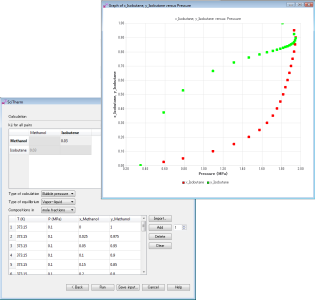
状態方程式の統計会合流体理論Statistical Associating Fluid Theory(SAFT) 1)およびその派生であるPC -SA FT 2)、e PC -SA FT 3)を利用して熱力学物性を推算するプログラムです。ポリマー、コポリマー、電解質等からなる混合物の物性値および相平衡を推算することができます。気液、液液、固液平衡、エネルギー(ギブスエネルギー、ヘルムホルツエネルギー)、エンタルピー、エントロピー、等温圧縮率、熱膨張係数、ジュールトムソン係数、音速、断熱体積弾性率等の物性を推算することができます。
- □対応プログラム
- SciTherm
薬物溶解度推算プログラム
PC-SAFT法により複数の純溶媒もしくは混合溶媒に対する薬品の溶解度を計算するためのインターフェースです。
- □対応プログラム
- SciPharma
構造活性相関プログラム
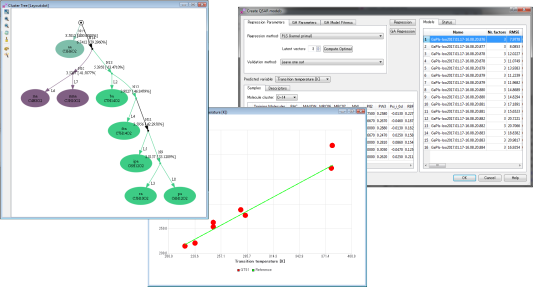
- □対応プログラム
- QSAR Plug-in
- ALVADESC DESCRIPTORS